Gene Sequencing
Introduction
In this school project I had the opportunity to implement dynamic programming algorithms that compute the minimal cost of aligning gene sequences and extract optimal alignments. These metrics can be used to interpret how similar two genes are and identify the best way to align them.
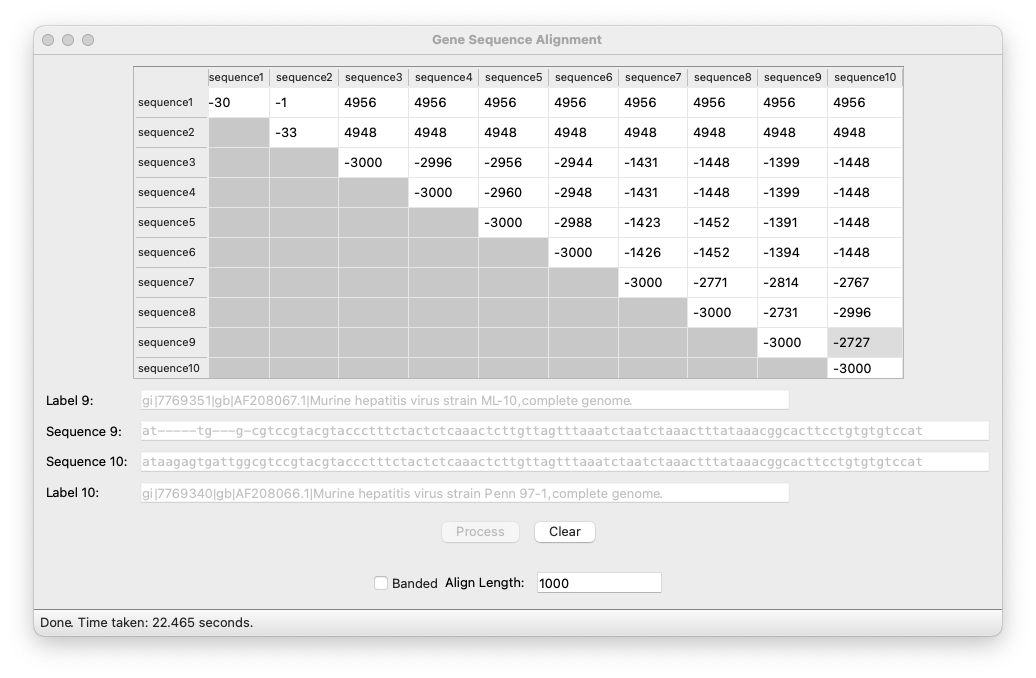
As input data we were given a number of DNA sequences, as well as some smaller testing strings. As output we displayed a matrix that showed how similar each sequence is to every other sequence. Clicking on each cell would display the selected sequences and their optimal alignment.
We also implemented a restricted ‘banded’ algorithm that yields faster, approximate results.
Unrestricted Approach
Dynamic programming is a process of solving problems using arrays. Generally, it means that the value for arr[i][j] is some mapping of arr[i-1][j], arr[i][j-1], and arr[i-1][j-1] (typically min). This way we can iteratively generate whatever final answer found at arr[n][m].
In this problem we used the Needleman/Wunsch cost function, which weights those array values:
- Substitutions - single character mis-matches, are penalized
1unit - Insertions/Deletions - ‘indels’, which create gaps, are penalized
5units - Matches are rewarded
-3units
So after initializing our array, we simply go row by row and replace the empty cells with min(arr[i-1][j] + 5, arr[i-1][j-1]-3, arr[i][j-1] + 5) if the previous characters match, or min(arr[i-1][j] + 5, arr[i-1][j-1]+1, arr[i][j-1] + 5) if they do not. This algorithm ensures that the final alignment string found avoids adding or deleting characters, prefering to substitute and match as much as it can.
The Unrestricted Algorithm had the requirement of running of 1000 base pairs in less than 120 seconds in O(nm) time and space.
Restricted ‘Banded’ Approach
The restricted approach is entirely like the unrestricted approach, except in how much of the array it uses. The banded approach specifies some parameter band width, which is the maximum number of used cells on each row. These bands ‘stair-case’ their way down towards the end of the 2D array, which results in faster computaton time, despite only achieving an approximation of the optimal result.
The Restricted Algorithm also had the requirement of running on 3000 base pairs in less than 10 seconds in $O(kn)$ time, where $k$ is the bandwidth constant and $n$ is the length of the shorter sequence.
Summary
As this was a school project I don’t want to show the whole repository I used, but here is some representational pseudo-code:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
# Used to implement Needleman-Wunsch scoring
MATCH = -3
INDEL = 5
SUB = 1
# ...
# Find the appropriate constant for Needleman Wunsch scoring on diagonal values.
MATCH_OR_SUB = MATCH if seq2[i - 1] == seq1[j - 1] else SUB
# Identify lowest cost
cost = MATCH_OR_SUB + table[i - 1][j - 1]
if (j != end_idx): # Evaluate value above
up_cost = INDEL + table[i - 1][j]
if up_cost <= cost:
cost = up_cost
if (j != start_idx): # Evaluate value left
left_cost = INDEL + table[i][j - 1]
if left_cost <= cost:
cost = left_cost
table[i][j] = cost
This project was awesome for learning Dynamic Programming. It’s definitley a really interesting approach to solving problems, albeit a tricky one to set up. The nice thing about debugging it however was that the transformation from state to state was so easy to follow. As soon as I knew that I was populating my table properly, most of the debugging had already handled itself.